
In the order phase, we used a whole-genome shotgun strategy to generate genome sequences of 45 new avian species. For 20 species, we produced high (>50x) coverage sequences from multiple libraries, with a gradient of insert sizes and built full-genome assemblies. For the remaining 25 species, we generated low (~30x) coverage data from two insert-size libraries and built less complete but still sufficient assemblies for comparative genome analyses.
In the family phase, we constructed pair-end libraries of one or two small insert sizes (250 bp, 280 bp, 500 bp, 800 bp) and one mate pair library (2 kb) for most samples. The read length for small insert libraries was 150 bp, and 49 bp for the mate pair library. The sequencing depth for most avian genomes ranged from 35x to 123x.
In the genus phase, we are applying a cobarcoding sequencing technology that enables the production of more contiguous genome assemblies for the genus-level analyses than were generated in the prior phase. This approach, called single-tube long-fragment read technology (stLFR), produces linked reads spanning long (>100 Kb) DNA fragments (Wang, Ou, et al. 2019). We generate 100X coverage stLFR data from high molecular weight DNA and use the data to generate a draft assembly with standardized computational pipelines. Tissue quality is of paramount importance. The poorer the quality, the shorter the DNA fragment length, and the worse the assembly quality. DNA with >40 kb fragments is best for stLFR sequencing although fragments ranging from 20 to 40 kb can be used to produce lower quality assemblies. One major benefit of the stLFR technology is that it requires relatively low starting amounts of DNA (1ng) than other approaches (>5 ug). For high quality tissue samples (e.g. frozen blood or muscle in liquid nitrogen or ethanol within 15 minutes of death) that yield long DNA fragments, we can produce assemblies with N50 scaffolds greater than 1Mb, and as high as 20 to 30Mb.
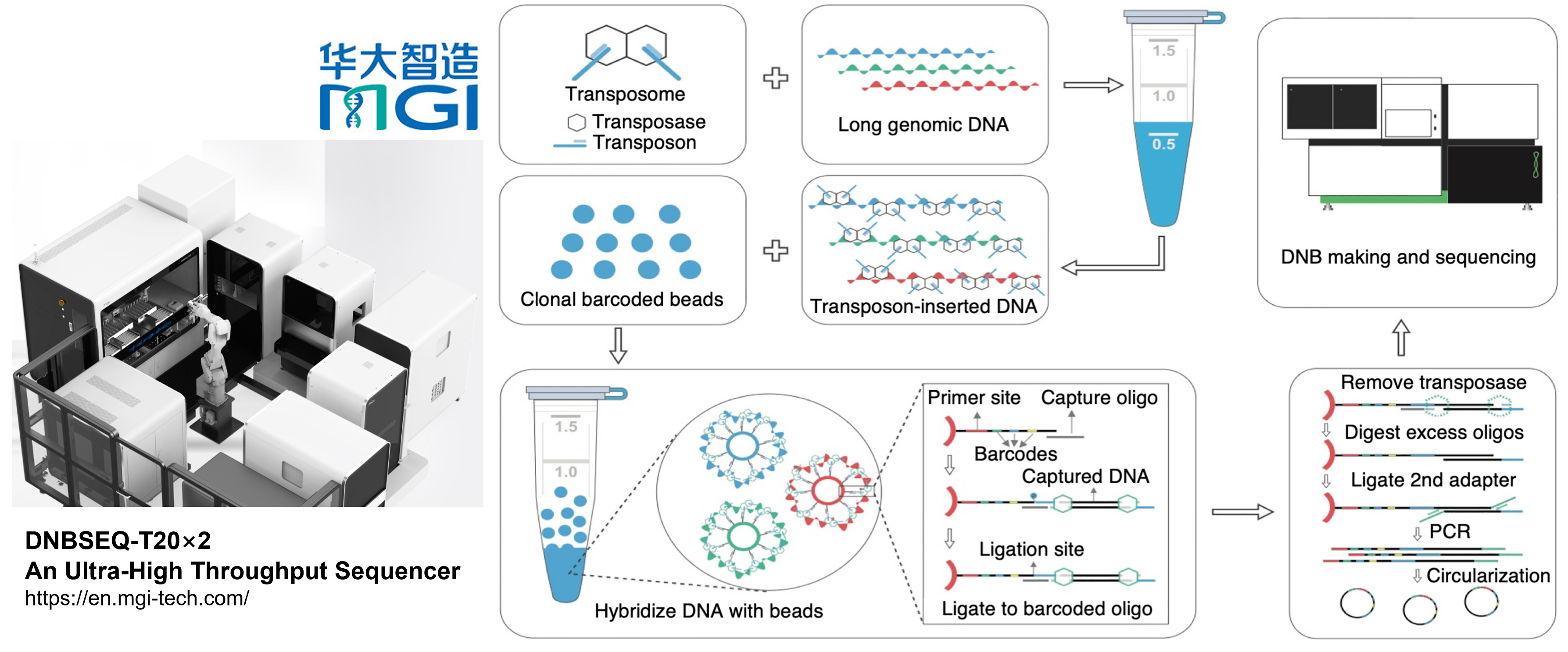
Overview of stLFR. The first step of stLFR involves inserting a hybridization sequence approximately every 200–1000 bp on long genomic DNA molecules. This is achieved using transposons. The transposon-integrated DNA is then mixed with beads that each contain ∼400,000 copies of an adapter sequence that contains a unique barcode shared by all adapters on the bead, a common PCR primer site, and a common capture sequence that is complementary to the sequence on the integrated transposons. After the genomic DNA is captured to the beads, the transposons are ligated to the barcode adapters. There are a few additional library processing steps and then the cobarcoded subfragments are sequenced on a BGISEQ-500 or equivalent sequencer.


The Bird 10,000 Genomes (B10K) Project is an initiative to generate representative draft genome sequences from all extant bird species. Based on the success of the previous ordinal level project , the project provided the first proof of concept in large-scale sequencing across a vertebrate class and a perspective on such discoveries that these genomes can make. The announcement of the B10K Project was published on 3rd June 2015 in Nature.
See About us page for contact info of B10K project organizers